2023.10.11
News
Detection and extraction of similar features in the disease-related gene groups ~Applying data-driven analysis on multiomics data~
Joint press release of Chuo University, Iwate Tohoku Medical Megabank Organization, and Iwate Medical University
Abstract
A research team composed of Professor Yoshihiro Taguchi of Chuo University and Iwate Tohoku Medical Megabank Organization (IMM) of Iwate Medical University, as a part of Tohoku Medical Megabank (TMM) Project1, applied an unsupervised data-driven analysis2, that was recently developed by Professor Taguchi, to the multiomics3 cohort datasets which were previously served as control datasets for case-control study, and extracted patterns shared across gene expression, DNA methylation4, and genetic variation. In addition, this team revealed that the data-driven analysis can extract various disease-related genes by identifying gene sets whose expression level varies with synchronizing to these patterns. They expect that further analyses of genes identified in this study will contribute to enabling the disease onset prediction. The research is published in PLOS ONE. (JST Aug.10, 2023).
Researchers
Yoshihiro H. TAGUCHI, Professor, Chuo University Faculty of Science and Engineering (Department of Physics)
Shohei KOMAKI, Lecturer, Institute for Biomedical Sciences of Iwate Medical University (Division of Biomedical Information Analysis)
Yoichi SUTOH, Associate Professor, Iwate Tohoku Medical Megabank Organization of Iwate Medical University (Division of Biomedical Information Analysis)
Hideki OHMOMO Associate Professor, Institute for Biomedical Sciences of Iwate Medical University (Division of Biomedical Information Analysis)
Yayoi YAMASAKI, Assistant Professor, Iwate Tohoku Medical Megabank Organization of Iwate Medical University (Division of Biomedical Information Analysis)
Atsushi SHIMIZU, Professor, Institute for Biomedical Sciences of Iwate Medical University (Division of Biomedical Information Analysis)
Published Journal: PLOS
Research Study
1. Background
Multiomics3 analysis that integrates different layers of profiles altogether is challenging, since the number of variables in profile substantially differ from each other. For instance, gene expression profile and genomic DNA methylation profile are often analyzed together, however, there are only tens of thousands of genes, whereas the number of DNA methylation sites are as many as tens of millions. The numbers differ one thousand times and the number of pairs between gene and DNA methylation sites are enormous. As such, it requires huge computational resources to conduct integrated analysis without controlling target numbers by focusing on DNA methylation sites in specific regions, such as promoter5 regions, based on prior knowledge. However, limiting the genomic regions to be analyzed, effects of DNA methylation on other regions (e.g., enhancer6) and functions remain unexplored
2 The Study and the Accomplishment
This study applied the method developed in the previous study* to treat multiomics data (gene expression profile, DNA methylation profile, Single Nucleotide Polymorphism (SNP)7 profile) which Iwate Tohoku Medical Megabank Organization (IMM) comprehensively collected from 100 local resident participants, and confirmed whether the relationships with disease-related gene can be identified or not. This is the data-driven approach called the variable extraction method which employed kernel tensor decomposition-based unsupervised study (hereafter called tensor decomposition), and this method is applicable to the datasets with all subjects belonging to a healthy group.
In addition, this method is implementable with kernel sized (square of the subject participants, in particular) or so memories per 1 profile, and thus, even for the enormous profiles such as genome and epigenome comprising tens of millions of SNP or DNA methylation sites, data-driven analysis can identify unique patterns across study subjects and identify the variables (gene expression profile, DNA methylation profile, SNP profile) that exhibit similarity to those patterns (Figure 1).
In this study, tensor decomposition was applied to multiomics data of each autosome retrieved from three cell types, CD4 positive T cells, monocytes, and neutrophils. As a result, the two patterns of subjects profiles were identified, and these two patterns of subjects observed in 22 autosomes show very strong mutual correlations between the other autosomes. As the genes identified in each autosome are completely independent from one another, it suggests that the observed patterns shared across chromosomes are not coincidence. The observed orthogonal patterns also cannot be explained by batch effects, and it is improbable that the same batch effects are present in the three omics profiles obtained by the different methodologies.
These two patterns of subjects are obtained as the second and the third singular value vectors by tensor decompositions, respectively. The second singular value vector was detected from all three cell types while the third singular value vector was detected from two cell types except monocytes. Then, the genes and DNA methylation regions with homological profiles with these patterns were selected to find out that these genes and regions are targeted by many transcription factors. Furthermore, the enrichment analysis revealed that these transcription factors relate to various diseases. In addition, the study found that identified SNP are statistically and significantly overlapped with binding sites of these transcription factors.
Therefore, the authors believe that the application of tensor decompositions is effective for the integrated analysis of multiomics datasets.
References:
*Taguchi, Yh., Turki, T. Novel feature selection method via kernel tensor decomposition for improved multiomics data analysis. BMC Med Genomics 15, 37 (2022). https://doi.org/10.1186/s12920-022-01181-4
3. Future Development
Besides the data used for this study, IMM accumulates umbilical blood data as well. IMM will continue accumulating multiomics data with an aim to establish, in the future, the means for national health management including the detection of pre-symptomatic state by analyzing multiomics data using tensor decompositions.
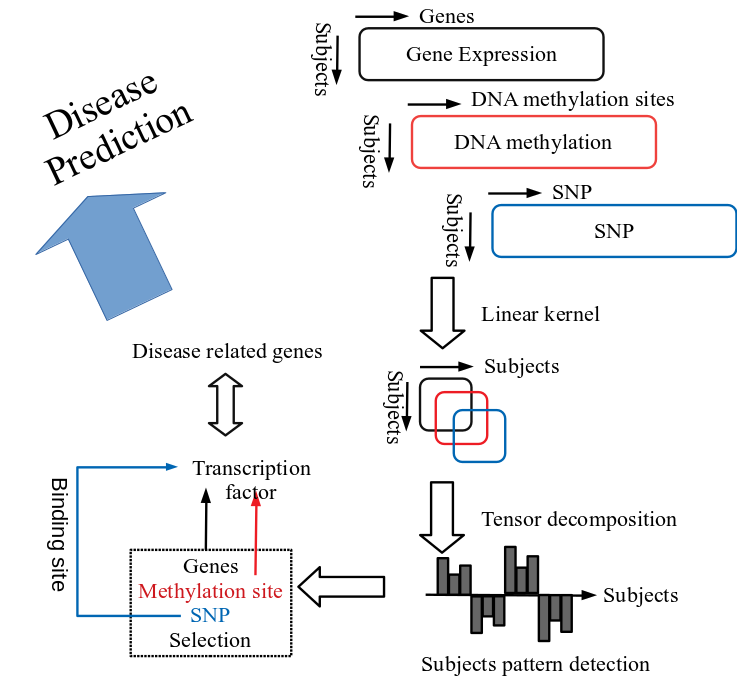
Figure 1. After converting Gene expression profile, DNA methylation profile, SNP profile into line of the subject participants’ square through linear kernelization to each, extract the subject pattern through tensor decompositions, and by selecting genes, DNA methylation sites, SNPs which synchronize with the patterns, we can select through the data-driven method without external information. Since many of the genes and DNA methylation sites are being targeted by transcription factors, and those transcription factors’ parts binding to DNA were statistically and significantly overlapped with SNP, integrated analysis of multiomics data can be said to be successful. In addition, considering the fact that transcription factors are related to disease, multiomics data plus tensor decompositions is a method of analysis which is expected to make future disease predictable (pre-symptomatic state).
This research was conducted with the support of Grant-in-Aid for Scientific Research (Ministry of Education, Culture, Sports, Science and Technology), Grant-in-Aid for Scientific Research on Innovative Areas (Research Area Proposal Type), 19H05270、20H04848, Fund for Grant-in-Aid for Scientific Research (Ministry of Education, Culture, Sports, Science and Technology), Grant-in-Aid for Scientific Research C, 20K12067.
Contact
-About the research-
Yoshihiro TAGUCHI
Professor of Faculty of Science and Engineering, Chuo University (Department of Physics)
E-mail: tagh★granular.com
Please replace ★ to @ when sending email.
-Inquiries-
Chuo University, Research Support Office
E-mail: kkouhou-grp★g.chuo-u.ac.jp
Please replace ★ to @ when sending email.
Glossary
1 Tohoku Medical Megabank (TMM) Project
Tohoku Medical Megabank Project was launched from FY2011 as a project to reconstruct from Great East Japan Earthquake, aiming to accomplish sound revitalization in the area as well as the implementations of customizations in disease prevention and medical. This project is initiated by Tohoku University Tohoku Medical Megabank Organization and Iwate Tohoku Medical Megabank Organization (IMM). In order to support the “Build Back Better” framework applied in medical systems at disaster area and to help the health-promotions of victims, the Organizations have conducted local residents’ cohort investigations and the third-generation cohort investigations to approximately 150 thousand residents so far since 2013, and it has been preparing Biobank by leveraging the retrieved samples/information.
Regarding the TMM Project, Japan Agency for Medical Research and Development (AMED) is now taking a role as an institution in charge of research support on this Project since 2015.
2Data-driven Usually, machine learning or data science require supervisions or enforced learnings to make model study in line with certain object. On the other hand, data-driven research do not set certain object but analyze the data at first. Then, taking the results in consideration, it observes the development in the data and lead to the conclusion. Such research method is called data-driven research method.
3Multiomics The coordinated term for DNA methylation profile or SNP profile excluding gene expression profile. Other than these, multiomics can be consisted from wide variety of profiles such as microRNA expression profile and histone-modifying.
4DNA methylation Among the 4 molecules that constitute human’s DNA (Adenine, thymine, guanine, cytosine), most of the cytosine is methylated. If the cytosine methylation happens, it influences gene expression by disturbing the binding of proteins which relates to transcriptional control (deter in general).
5Promoter Among DNA that constitute genome, gene function area occupy just a small portion. In order to decide which gene to express in appropriate time and appropriate organization, the area adjacent to gene is material, and such area is called “promoter.” The expression amount varies if this area becomes methylated or newly mutated.
6Enhancer, Silencer Unlike Promoter, there is also an area far from gene which affect gene expression through DNA loop structure formation. Among this, the area which increase the expression is called enhancer, and the area which control the expression is called silencer. The DNA methylation or newly mutation on this area also have effects on gene expression.
7Single Nucleotide Polymorphism (SNP) A location that one of the 4 kinds of nucleotides may have been replaced to another molecule, depending on individual. Since protein within an organism is made by lining up 20 kinds of amino acid in DNA sequence, the structure of protein change and its function may have a significant impact depending on DNA position where SNP exists.
8Transcription factor The collective term for protein group which controls gene expression. The gene expression is controlled when transcription factor is binding to promoter/enhancer area.
9Kernelization (kernel trick) A mathematical trick that analyzes very high dimension space with few sample. It is routinely used in machine learning.
